Descifran el genoma de Fusarium chlamydosporum, un hongo que amenaza los cultivos de zarzamora en México
AUTORES
Anton Pashkova 1, José Pedro Martínez-Hernández 2, Alfredo Herrera-Estrella 2, 6 , Pablo Cruz-Morales 3 , 6, Nelly Selem-Mojica 5, 6 , José Manuel Villalobos-Escobedo 4 ,6
Escuela Nacional de Estudios Superiores unidad Morelia, Universidad Nacional Autónoma de México, Morelia, Mexico.1
Unidad de Genómica Avanzada, Cinvestav, 36824 Irapuato, Guanajuato. Mexico.2
Yeast Natural Products, The Novo Nordisk Foundation Center for Biosustainability, Technical University of Denmark, Denmark.3
Tecnológico de Monterrey, Institute for Obesity Research, Ave. Eugenio Garza Sada 2501, Monterrey, N.L., 64849, Mexico.4
Centro de Ciencias Matemáticas, Universidad Nacional Autónoma de México, Morelia, Mexico.5
The LatAmBio Initiative, Irapuato, Guanajuato, Mexico.6
e-mail: jose.villalobos@tec.mx ; nselem@matmor.unam.mx
Descarga el artículo
En Michoacán —uno de los principales productores de berries del país—, los agricultores han enfrentado pérdidas severas por un enemigo invisible:
Fusarium, el hongo responsable de la marchitez y muerte de plantas completas.
En este estudio, se logró aislar e identificar la cepa causante de la infección en zarzamoras mexicanas, denominada IraGTOF6, perteneciente a la especie Fusarium chlamydosporum.
Este hallazgo es especialmente relevante porque el patógeno más común suele ser F. oxysporum; sin embargo, el aislamiento de una especie diferente sugiere que nuevas variantes del hongo están adaptándose a los suelos locales, lo que podría complicar su control en el futuro.
Para entender su comportamiento, el equipo de investigación realizó el ensamblaje híbrido más completo hasta ahora del genoma de F. chlamydosporum, combinando tecnologías de secuenciación Illumina y Oxford Nanopore.
Esta divergencia genética sugiere la presencia de mecanismos de virulencia únicos que podrían explicar su resistencia al suelo alcalino y su alta virulencia en cultivos de zarzamora.
Más allá del avance científico, el estudio tiene un impacto directo en la agricultura mexicana: los datos generados servirán para diseñar biomarcadores moleculares que permitan detectar la enfermedad antes de que cause pérdidas, y para desarrollar estrategias de control biológico más sostenibles, reduciendo la dependencia de fungicidas químicos.
El genoma de Fusarium chlamydosporum IraGTOF6 se convierte así en una herramienta estratégica para mejorar la sanidad vegetal, proteger la producción nacional de zarzamora y fortalecer la seguridad alimentaria desde la ciencia mexicana.

Fig 1: Es muy probable que IraGTOF6 pertenezca a Fusarium chlamydosporum según el análisis filogenético y los resultados de BLAST. 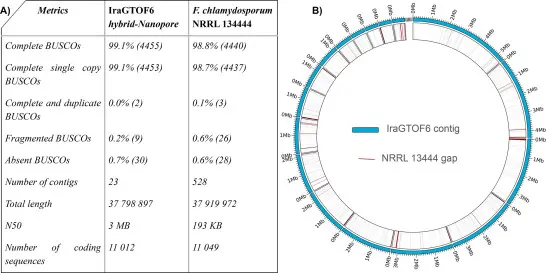
Fig 2: El ensamblaje híbrido de IraGTOF6 muestra mejoras significativas en contigüidad e integridad sobre NRRL13444. 
Fig 3: Análisis filogenómico de Fusarium basado en el núcleo del proteoma, que colocó a IraGTOF6 junto a la especie F. Chlamydosporum
- H.S. Buttar, A. Singh, A. Sirari, Kaur K Anupam, A. Kumar, et al.Investigating the impact of fungicides and mungbean genotypes on the management of pod rot disease caused by Fusarium equiseti and Fusarium chlamydosporumFront. Plant Sci., 14 (2023)Google Scholar
- [2]T.J. Parihar, M.Y. Sofi, R.S. Rasool, S. Khursheed, Z.A. Bhat, K. Hussain, et al.Fusarium chlamydosporum, causing wilt disease of chili (Capsicum annum L.) and brinjal (Solanum melongena L.) in Northern Himalayas: a first reportSci. Rep., 12 (1) (2022), Article 20392View in ScopusGoogle Scholar
- [3]B.H. Segal, T.J. Walsh, J.M. Liu, J.D. Wilson, K.J. Kwon-ChungInvasive infection with fusarium chlamydosporum in a patient with aplastic anemiaJ. Clin. Microbiol., 36 (6) (1998), pp. 1772-1776CrossrefView in ScopusGoogle Scholar
- [4]A. Burkhardt, P.M. Henry, S.T. Koike, T.R. Gordon, F. MartinDetection of fusarium oxysporum f. sp. fragariae from infected strawberry plantsPlant Dis., 103 (5) (2019), pp. 1006-1013CrossrefView in ScopusGoogle Scholar
- [5]N. Saitou, M. NeiThe neighbor-joining method: a new method for reconstructing phylogenetic treesMol. Biol. Evol., 4 (4) (1987), pp. 406-425View in ScopusGoogle Scholar
- [6]Krueger F., James F., Ewels P., Afyounian E., Weinstein M., Schuster-Boeckler B., et al. FelixKrueger/TrimGalore: v0.6.10 Zenodo; 2023. https://doi.org/10.5281/zenodo.7598955Google Scholar
- [7]R.R. Wick, L.M. Judd, C.L. Gorrie, K.E. HoltCompleting bacterial genome assemblies with multiplex MinION sequencingMicrob. Genomics, 3 (10) (2017), Article e000132Google Scholar
- [8]W. De Coster, R. RademakersNanoPack2: population-scale evaluation of long-read sequencing dataBioinformatics, 39 (5) (2023), p. btad311View in ScopusGoogle Scholar
- [9]A.V. Zimin, G. Marçais, D. Puiu, M. Roberts, S.L. Salzberg, J.A. YorkeThe MaSuRCA genome assemblerBioinformatics, 29 (21) (2013), pp. 2669-2677CrossrefView in ScopusGoogle Scholar
- [10]D. Guan, S.A. McCarthy, J. Wood, K. Howe, Y. Wang, R. DurbinIdentifying and removing haplotypic duplication in primary genome assembliesBioinformatics, 36 (9) (2020), pp. 2896-2898CrossrefView in ScopusGoogle Scholar
- [11]M.C. FrithA new repeat-masking method enables specific detection of homologous sequencesNucleic. Acids. Res., 39 (4) (2011), p. e23CrossrefView in ScopusGoogle Scholar
- [12]J.M. Flynn, R. Hubley, C. Goubert, J. Rosen, A.G. Clark, C. Feschotte, A.F. SmitRepeatModeler2 for automated genomic discovery of transposable element familiesProc. Natl. Acad. Sci. U.S.A., 117 (17) (2020), pp. 9451-9457CrossrefView in ScopusGoogle Scholar
- [13]M. Manni, M.R. Berkeley, M. Seppey, F.A. Simão, E.M. ZdobnovBUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomesMol. Biol. Evol., 38 (10) (2021)4647-54.14Google Scholar
- [14]J.M. Palmer, J. StajichFunannotate v1.8.1: eukaryotic genome annotation Zenodohttps://zenodo.org/records/4054262 (2020)Google Scholar
- [15]M. Stanke, S. WaackGene prediction with a hidden Markov model and a new intron submodelBioinformatics, 19 (suppl_2) (2003)ii215-25Google Scholar
- [16]The UniProt ConsortiumUniProt: the Universal protein knowledgebase in 2025Nucleic Acids Res., 53 (D1) (2025), pp. D609-D617Google Scholar
- [17]P. Jones, D. Binns, H.Y. Chang, M. Fraser, W. Li, C. McAnullaInterProScan 5: genome-scale protein function classificationBioinformatics, 30 (9) (2014), pp. 1236-1240CrossrefView in ScopusGoogle Scholar
- [18]K. Blin, S. Shaw, A.M. Kloosterman, Z. Charlop-Powers, G.P. van Wezel, M.H. Medema, T. WeberantiSMASH 6.0: improving cluster detection and comparison capabilitiesNucleic. Acids. Res., 49 (W1) (2021), pp. W29-W35CrossrefView in ScopusGoogle Scholar
- [19]C.P. Cantalapiedra, A. Hernández-Plaza, I. Letunic, P. Bork, J. Huerta-CepaseggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scaleMol. Biol. Evol., 38 (12) (2021), pp. 5825-5829CrossrefView in ScopusGoogle Scholar
- [20]R.C. EdgarSearch and clustering orders of magnitude faster than BLASTBioinformatics, 26 (19) (2010), pp. 2460-2461View at publisherCrossrefView in ScopusGoogle Scholar
- [21]N.M. Chaudhari, V.K. Gupta, C. DuttaBPGA- an ultra-fast pan-genome analysis pipelineSci. Rep., 6 (2016), Article 24373View in ScopusGoogle Scholar
- [22]B.Q. Minh, H.A. Schmidt, O. Chernomor, D. Schrempf, M.D. Woodhams, A. von Haeseler, et al.IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic eraMol. Biol. Evol., 37 (5) (2020), pp. 1530-1534View at publisherCrossrefView in ScopusGoogle Scholar
- [23]S. Kalyaanamoorthy, B.Q. Minh, T.K.F. Wong, A. von Haeseler, L.S JermiinModelFinder: fast model selection for accurate phylogenetic estimatesNat. Methods, 14 (6) (2017), pp. 587-589View at publisherCrossrefGoogle Scholar